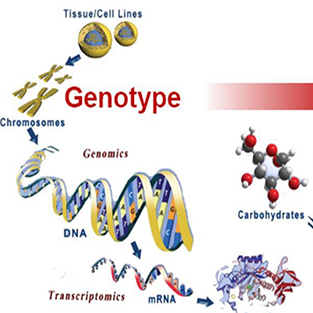
The types and quantities of metabolites vary greatly in different species and tissues. The mGWAS (metabolome Genome-Wide Association Study) approach uses population materials, based on resequencing genetic information and metabolomics data to carry out genome-wide association analysis between genomic variations and metabolite quantities.
Case Study: Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism (Chen et al., Nature Genetics 2014)
Subspecies specific resequencing and metabolomics between two rice subspecies
Using 529 rice leaf materials, a total of 840 compounds were detected by Widely-Targeted Metabolomics. Cluster analysis of these metabolites identified C-glycosylated and malonylated flavonoids accumulated more in the O. indica, while the O. japonica contained large amounts of phenolamides and arabidopyl derivative compounds. These subspecies-specific metabolites reflect the differentiation of O. indica and O. japonica.
SNP analysis and metabolite association of the rice population
Using NGS on the 529 materials, 6,428,770 SNPs were screened for further analysis. Simple linear regression (LR) and linear mixed models (LMM) were used for mGWAS analysis and a total of 2,947 major SNPs were identified, corresponding to 634 loci in at least one population.
161 significant loci shown on the Manhattan plot correspond to flavonoids, phenolic compounds, amino acids and their derivatives, terpenoids, nucleic acids and their derivatives, and other known metabolites. In addition, there are 195 loci corresponding to unknown metabolites.
mGWAS assists with annotation of unknown metabolites
mGWAS facilitates the annotation of metabolites by associating unknown metabolites with functionally relevant genes. For example, SNP sf1207801034 in the NOMT gene encoding naringenin 7-O-methyltransferase correlated with mr1002 levels, suggesting that this metabolite may be sakuranetin. Subsequently, the mr1002 metabolite was proven to be sakuranetin by comparing the retention time and fragmentation pattern with the chemical standard of this compound. In this study, GWAS was able to annotate 166 unknown metabolites.
Biochemical and functional interpretation of GWAS results
In addition to providing insight into the genetic basis of metabolic changes, mGWAS also provide insight into the biochemistry and function of underlying pathways. We can examine previously unidentified candidate genes by (i) finding genes or gene clusters associated with the relevant metabolic signatures encoded by these loci; (ii) clustering candidate genes with homologous genes with known functions; (iii) cross-reference genetic map results. We hope to uncover candidate genes or SNPs valuable for plant physiological or human nutrition.
For instance, trigonelline, an N-methyl conjugate of nicotinic acid, has long been reported to regulate various processes, especially abiotic stress in plants, but the enzyme that catalyzes niacin to trigonelline was unknown at the time. Trigonelline levels were significantly correlated with SNP sf0235317720 on chromosome 2, which was in linkage disequilibrium with the Os02g57760 gene encoding an O-methyltransferase protein, indicating that Os02g57760 encodes a key methyltransferase that catalyzes trigonelline biosynthesis. When N-terminal His-tagged Os02g57760 was expressed in E. coli BL-21, we were able to detect niacin: N-methyltransferase activity in soluble protein extracts. Consistent with the in vitro activity, overexpression of Os02g57760 in ZH11 (an O. japonica cultivar with low trigonelline content) resulted in a greater accumulation of trigonelline in the transgenic lines compared to control plants, confirming that Os02g57760 catalyzes trigonelline biosynthesis.
For more information about metabolome project, please feel free to contact us!
Ubicación : Building C2-3 No. 666 Gaoxin Avenue, East Lake New Technology Development Zone, Wuhan, 430073 wuhan,
Persona a contactar : bio metware, 027 65520430